Seznamy 36+ Rdkit Atom Index
Seznamy 36+ Rdkit Atom Index. Each atom maintains a dict of properties:. Many of the methods of atom require that the atom be associated with a molecule (an romol).; > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …

Nejchladnější Rdkit Macs In Chemistry
The _smilesatomoutputorder property used to be the only way to get a canonical ordering. Each property is keyed by name and can store an arbitrary type.; The degree is independent of bond orders, but is dependent. Many of the methods of atom require that the atom be associated with a molecule (an romol).; This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted.The class for representing atoms.
Summary files reviews support wiki mailing lists code. The degree is independent of bond orders, but is dependent. Summary files reviews support wiki mailing lists code. Each atom maintains a dict of properties:. Each property is keyed by name and can store an arbitrary type.; The _smilesatomoutputorder property used to be the only way to get a canonical ordering.

> i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The degree is independent of bond orders, but is dependent. Each property is keyed by name and can store an arbitrary type.; This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of ….. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule.

The _smilesatomoutputorder property used to be the only way to get a canonical ordering... > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule.

Returns the degree of the atom in the molecule... However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … The degree is independent of bond orders, but is dependent. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. The class for representing atoms. Summary files reviews support wiki mailing lists code. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.
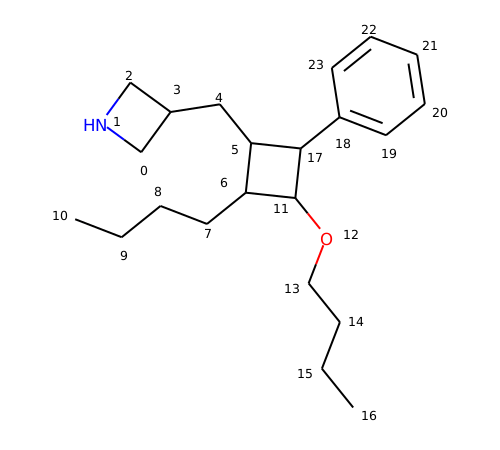
Many of the methods of atom require that the atom be associated with a molecule (an romol).; . The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.

That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5... That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Each atom maintains a dict of properties:. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted.

Many of the methods of atom require that the atom be associated with a molecule (an romol).; The degree is independent of bond orders, but is dependent. The class for representing atoms.. The _smilesatomoutputorder property used to be the only way to get a canonical ordering.

The degree is independent of bond orders, but is dependent... . Summary files reviews support wiki mailing lists code.

Each property is keyed by name and can store an arbitrary type.;. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Returns the degree of the atom in the molecule. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Each atom maintains a dict of properties:. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Summary files reviews support wiki mailing lists code.

Returns the degree of the atom in the molecule... That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Summary files reviews support wiki mailing lists code. Each atom maintains a dict of properties:. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int :

Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Each atom maintains a dict of properties:. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Returns the degree of the atom in the molecule.

Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. The class for representing atoms. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Many of the methods of atom require that the atom be associated with a molecule (an romol).; At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Returns the degree of the atom in the molecule. Summary files reviews support wiki mailing lists code.. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int :

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int :

Each property is keyed by name and can store an arbitrary type.;.. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : The degree is independent of bond orders, but is dependent. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Each atom maintains a dict of properties:.

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. The class for representing atoms. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Many of the methods of atom require that the atom be associated with a molecule (an romol).; > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule.. Many of the methods of atom require that the atom be associated with a molecule (an romol).;

Summary files reviews support wiki mailing lists code. Summary files reviews support wiki mailing lists code. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. Each property is keyed by name and can store an arbitrary type.; At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Each atom maintains a dict of properties:. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. The degree is independent of bond orders, but is dependent. Returns the degree of the atom in the molecule. The class for representing atoms. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Summary files reviews support wiki mailing lists code.

Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int :. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences.. The _smilesatomoutputorder property used to be the only way to get a canonical ordering.

Returns the degree of the atom in the molecule. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Returns the degree of the atom in the molecule. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. The class for representing atoms. Each atom maintains a dict of properties:. Each property is keyed by name and can store an arbitrary type.; However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …

Many of the methods of atom require that the atom be associated with a molecule (an romol).;.. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Each property is keyed by name and can store an arbitrary type.; Each atom maintains a dict of properties:. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. The class for representing atoms. The degree is independent of bond orders, but is dependent. The _smilesatomoutputorder property used to be the only way to get a canonical ordering... However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …

Each property is keyed by name and can store an arbitrary type.; However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Each atom maintains a dict of properties:.. Each atom maintains a dict of properties:.

Each atom maintains a dict of properties:. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. The class for representing atoms. Summary files reviews support wiki mailing lists code. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.

The _smilesatomoutputorder property used to be the only way to get a canonical ordering... That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Each atom maintains a dict of properties:. Each property is keyed by name and can store an arbitrary type.; The class for representing atoms. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule.. The degree is independent of bond orders, but is dependent.

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Summary files reviews support wiki mailing lists code. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Each property is keyed by name and can store an arbitrary type.; That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. The class for representing atoms. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering.. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.
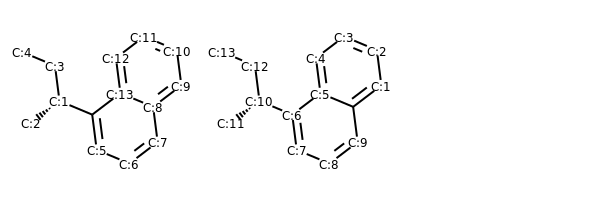
Each property is keyed by name and can store an arbitrary type.; The _smilesatomoutputorder property used to be the only way to get a canonical ordering. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Returns the degree of the atom in the molecule. Summary files reviews support wiki mailing lists code. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Each property is keyed by name and can store an arbitrary type.; Each atom maintains a dict of properties:. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5.. The _smilesatomoutputorder property used to be the only way to get a canonical ordering.

That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.

The _smilesatomoutputorder property used to be the only way to get a canonical ordering... Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.
It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Returns the degree of the atom in the molecule. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering.. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.

Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : The _smilesatomoutputorder property used to be the only way to get a canonical ordering. The degree is independent of bond orders, but is dependent. The class for representing atoms. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering.

This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Summary files reviews support wiki mailing lists code. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.. Returns the degree of the atom in the molecule.

The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule... That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. The degree is independent of bond orders, but is dependent... The degree is independent of bond orders, but is dependent.

Returns the degree of the atom in the molecule.. Summary files reviews support wiki mailing lists code. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.

At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences.. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : Each property is keyed by name and can store an arbitrary type.; Many of the methods of atom require that the atom be associated with a molecule (an romol).; The class for representing atoms. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Each atom maintains a dict of properties:. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int :

This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Each atom maintains a dict of properties:. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The degree is independent of bond orders, but is dependent... At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences.

> i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Each atom maintains a dict of properties:.. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering.

The degree is independent of bond orders, but is dependent. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. The class for representing atoms. The degree is independent of bond orders, but is dependent. Summary files reviews support wiki mailing lists code. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Each atom maintains a dict of properties:. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5.

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of ….. Each atom maintains a dict of properties:.

Each atom maintains a dict of properties:. Each atom maintains a dict of properties:. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Summary files reviews support wiki mailing lists code. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. The degree is independent of bond orders, but is dependent. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. The class for representing atoms. Many of the methods of atom require that the atom be associated with a molecule (an romol).; It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering... Each property is keyed by name and can store an arbitrary type.;
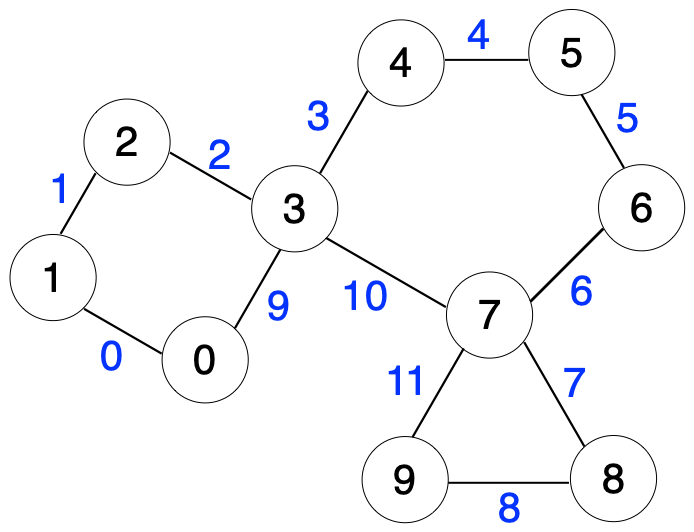
The _smilesatomoutputorder property used to be the only way to get a canonical ordering.. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Returns the degree of the atom in the molecule.. Each property is keyed by name and can store an arbitrary type.;

Each property is keyed by name and can store an arbitrary type.; That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Summary files reviews support wiki mailing lists code. The degree is independent of bond orders, but is dependent. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Returns the degree of the atom in the molecule. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Each property is keyed by name and can store an arbitrary type.; > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule.
Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Each atom maintains a dict of properties:. Returns the degree of the atom in the molecule. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Each property is keyed by name and can store an arbitrary type.;. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule.

Many of the methods of atom require that the atom be associated with a molecule (an romol).; > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Returns the degree of the atom in the molecule. The degree is independent of bond orders, but is dependent. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Summary files reviews support wiki mailing lists code. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences.

Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.. The class for representing atoms. Each property is keyed by name and can store an arbitrary type.; It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Summary files reviews support wiki mailing lists code.

However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Each atom maintains a dict of properties:. Returns the degree of the atom in the molecule. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : The class for representing atoms. The class for representing atoms.
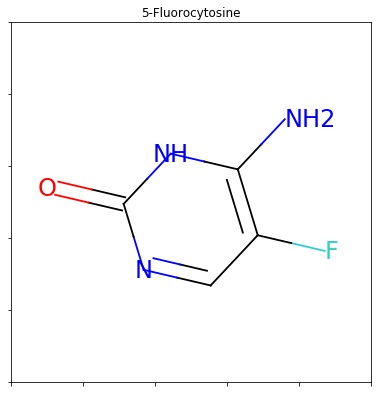
It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering... At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. The class for representing atoms. Each property is keyed by name and can store an arbitrary type.; It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Returns the degree of the atom in the molecule. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.

Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Summary files reviews support wiki mailing lists code.

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering... Each atom maintains a dict of properties:. The degree is independent of bond orders, but is dependent. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Many of the methods of atom require that the atom be associated with a molecule (an romol).;

This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted.. The class for representing atoms. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Many of the methods of atom require that the atom be associated with a molecule (an romol).; At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : The degree is independent of bond orders, but is dependent. Each atom maintains a dict of properties:... Many of the methods of atom require that the atom be associated with a molecule (an romol).;

The degree is independent of bond orders, but is dependent... Each atom maintains a dict of properties:. Summary files reviews support wiki mailing lists code. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. Many of the methods of atom require that the atom be associated with a molecule (an romol).;. Returns the degree of the atom in the molecule.

Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called... > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Returns the degree of the atom in the molecule. Summary files reviews support wiki mailing lists code. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering... The class for representing atoms. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Returns the degree of the atom in the molecule.

The _smilesatomoutputorder property used to be the only way to get a canonical ordering.. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Returns the degree of the atom in the molecule.. Summary files reviews support wiki mailing lists code.

However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of ….. Each property is keyed by name and can store an arbitrary type.; This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. The class for representing atoms. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Returns the degree of the atom in the molecule. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Summary files reviews support wiki mailing lists code... The _smilesatomoutputorder property used to be the only way to get a canonical ordering.

This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Returns the degree of the atom in the molecule. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. Each atom maintains a dict of properties:.. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …

This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. Returns the degree of the atom in the molecule. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Each atom maintains a dict of properties:. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int :.. The class for representing atoms.

The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : The degree is independent of bond orders, but is dependent. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. The class for representing atoms. Each atom maintains a dict of properties:. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5.

> i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. The degree is independent of bond orders, but is dependent. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.. Returns the degree of the atom in the molecule.

This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. The class for representing atoms. Each property is keyed by name and can store an arbitrary type.; That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Each atom maintains a dict of properties:. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5.
The degree is independent of bond orders, but is dependent. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. The degree is independent of bond orders, but is dependent. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Returns the degree of the atom in the molecule. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.. Returns the degree of the atom in the molecule.

Each atom maintains a dict of properties:... Summary files reviews support wiki mailing lists code. Many of the methods of atom require that the atom be associated with a molecule (an romol).; The class for representing atoms. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. The degree is independent of bond orders, but is dependent. Each property is keyed by name and can store an arbitrary type.;

Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Each atom maintains a dict of properties:. The class for representing atoms. The _smilesatomoutputorder property used to be the only way to get a canonical ordering. Each property is keyed by name and can store an arbitrary type.; However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Returns the degree of the atom in the molecule. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences.. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called.

The _smilesatomoutputorder property used to be the only way to get a canonical ordering.. Summary files reviews support wiki mailing lists code. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of … Each property is keyed by name and can store an arbitrary type.; At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted.. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5.
Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5.
Many of the methods of atom require that the atom be associated with a molecule (an romol).; Returns the degree of the atom in the molecule. The class for representing atoms. Each property is keyed by name and can store an arbitrary type.; Summary files reviews support wiki mailing lists code. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. The degree is independent of bond orders, but is dependent.. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering.
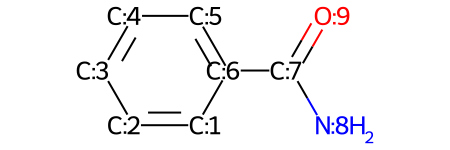
The class for representing atoms.. Rdkit::atom::chiraltype getchiraltag (rdkit::atom {lvalue}) getdegree((atom)arg1) → int : The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. > i have fragmented a molecule using getmolfrags and want to relate the atoms in the fragments to the original molecule. Each property is keyed by name and can store an arbitrary type.; Each atom maintains a dict of properties:. The degree is independent of bond orders, but is dependent. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Returns the degree of the atom in the molecule. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering... The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.

The class for representing atoms... Each atom maintains a dict of properties:. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering.. The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule.
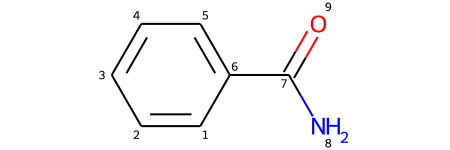
The getidx () method seems to be retrieving atomic indexes way beyond those that should be in the created molecule. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences. That is, there is no rotation of the molecule (that i can see) that will allow you to superimpose f0, h3, h4, and h5. Returns the degree of the atom in the molecule. It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Properties can be marked as calculated, in which case they will be cleared when the clearcomputedprops() method is called. Many of the methods of atom require that the atom be associated with a molecule (an romol).;.. However, i encountered a problem that rdkit.chem.rdmolops.getadjacencymatrix() doesn't provide the index of …

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering. Many of the methods of atom require that the atom be associated with a molecule (an romol).; Each property is keyed by name and can store an arbitrary type.; It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering.. Returns the degree of the atom in the molecule.

It is still useful if you care about the order of atoms in the output smiles, but is quite inefficient if all you want is a canonical ordering... Returns the degree of the atom in the molecule. The class for representing atoms. Many of the methods of atom require that the atom be associated with a molecule (an romol).; This seems to occur randomly, as multiple attempts on the same reaction block show that it may indeed be converted.. At the same time, i'm not sure whether rdkit is super strict about adhering the daylight smiles spec, or if it has known differences.


